
The head chair of pathology and a graduate research assistant from the University of Tennessee Health Science Center detail their paper published in Oncotarget titled, “The critical role that STAT3 plays in glioma-initiating cells: STAT3 addiction in glioma.”
The Behind the Study series transcribes videos of chosen researchers elaborating on their recent papers published in Oncotarget. Visit the Oncotarget YouTube channel for more insights from outstanding authors.
—
Lawrence Pfeffer
Hi, I’m Lawrence Pfeffer. I’m the head chair of pathology at the University of Tennessee Health Science Center. And we’re going to talk about the publication entitled The Critical Role That STAT3 Plays in Glioma-initiating Cells: STAT3 Addiction in Glioma. And this work was done mainly by my graduate student who is sitting next to me, who will introduce herself right afterwards, and some members of my lab, Meiyun Fan, Chuan He Yang, pathologist who we work with, Blazej Zbytek, and then two of our colleagues over at St. Jude who played some important critical roles in some of the studies in gene expression analysis. And that was David Finkelstein and Martine Roussel over at St. Jude’s. And I’m just going to introduce why we started on this project.
My lab has been interested in the STAT3 transcription factor for many years, dating back to my time when I was in New York City and we were looking at interferon signaling and we had identified that STAT3 was activated by the cytokine interferon. And for a long time, I wondered if STAT3, since it played such an important role in regulating cell growth and a number of biologic properties, if it was also involved in cancer. And a number of publications came out around 20 years ago showing that there was constitutive activation of STAT3 in a number of different cancers. And I had an interest in glioma for a number of years due to the fact that I had a close colleague who unfortunately came down with glioblastoma and died shortly after diagnosis. So I always had, in the back of my mind, an interest in studying this devastating form of brain cancer.
So in a previous iteration of this project, with a former graduate student, we studied in glioblastoma cancer stem cells, if STAT3 was constitutively active as measured by its tyrosine phosphorylation STAT3 inhibitor in a mouse study, a subcutaneous mouse study, and showed that a STAT3 phosphorylation inhibitor blocked the growth of the tumors. So Debolina, who we’ll talk next, came to me, came to work in my lab. And I said, “It would be great if we could piece out the different phosphorylation sites within the STAT3 protein and define what do they do specifically in regulating glioblastoma develop and focusing specifically on glioblastoma cancer stem cells,” figuring that this would be rather straightforward project. But as Debolina will smirk and tell you, we found that it was not as simple a project, but still, in the end, she got some really exciting results. So I’ll let Debolina introduce herself and take over.
Debolina Ganguly
Hello everyone. My name is Debolina Ganguly. I’m a graduate research assistant under Dr. Lawrence Pfeffer at the University of Tennessee Health Science Center located in Memphis, Tennessee. So the paper that Dr. Pfeffer talked about was my PhD project. And like Dr. Pfeffer mentioned, we started this project at the time when we had just published a paper using STAT3 inhibitors in glioma cancer stem-like cells. And we found that using those inhibitors led to an inhibition of tumorigenesis. Now, STAT3 has multiple phosphorylation sites, but the two most critical sites are the tyrosine 705 and the serine 727. So we became interested in investigating the role of these two phosphorylation sites, and so I had to knock out the expression of STAT3 in the cancer stem-like cells and then investigate the role of the two phosphorylation sites of STAT3.
Now, as it’s done in the field, studying any gene expression, we do it by CRISPR-Cas9 knock out. That didn’t work in the cancer stem-like cells in that these cells are so addicted to STAT3 that knocking out STAT3 made the cells die. I then tried to do a constitute of knock down of STAT3, and that didn’t work either. So the only system that worked in these cells is the inducible knockdown system. I was able to establish the inducible knockdown system in vitro, and then later on. And interesting finding of this study was that in the cancer stem-like cells, I found that the tyrosine phosphorylation site of STAT3 is critical for STAT3 induced GIC tumorigenesis.
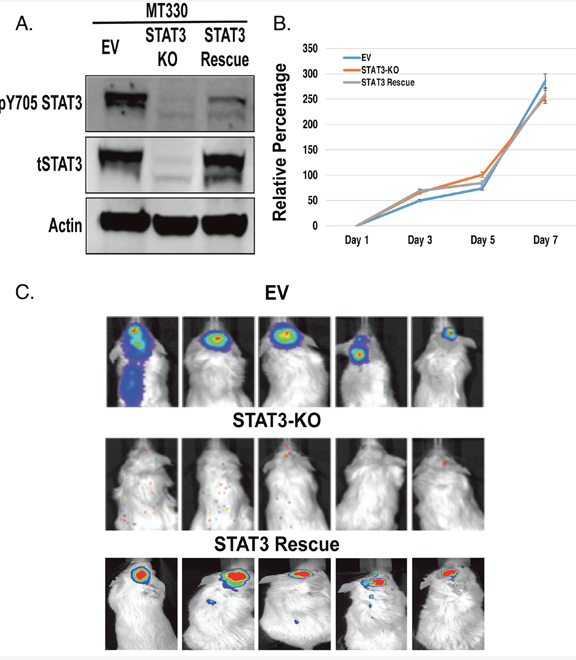
The serine 727 site in the cancer stem-like cells of GBM was dependent on tyrosine 705 phosphorylation. And I say that because in GBM cell lines such as MT330, we had established the system also in those cells, and in those cells we found that serine 727 phosphorylation happened without tyrosine 705 phosphorylation, but in the cancer stem-like cells, serine phosphorylation was dependent on 705 tyrosine phosphorylation.
So we were able to establish the phosphorylation system in the inducible STAT3 knockdown background. And then we did some high throughput gene expression analysis, and we found that STAT3 regulates a bunch of genes. We found close to …
Lawrence Pfeffer
… 150 genes.
Debolina Ganguly
150 genes that were regulated by STAT3. And what was interesting is we found that a bunch of genes which regulate the extra cellular matrix are regulated by STAT3, and these included genes, collagen genes, then matrix metalloproteinases. We found a bunch of interferon related genes, which are regulated by STAT3. And future studies will be directed towards addressing these genes, which seem to be STAT3 regulated in the cancer stem-like cells. So that is a gist of what we tried to do in this paper.
Lawrence Pfeffer
I think it was a shock that STAT3, the phosphorylation on the serine site was so completely dependent on the tyrosine site. And Debolina kind of glossed over of what a technically difficult thing this was because we took the standard glioblastoma line that we had growing in the lab and we were able to edit it out by CAS CRISPR gene editing. We took out the STAT3 gene, and when we put those STAT3 knock out glioma cells into mice, they didn’t form tumors at all, which was a real surprising result. We expected kind of a, maybe a slight inhibition, but we never got any tumors formed. And we knew that it was really highly STAT3 dependent because if you put stat Dre back into the knock out cells, you got tumorigenesis.
So we thought this would be such a straightforward process when we moved to the cancer stem cells. And it was just really surprising that first, we tried the CAS CRISPR knock out, didn’t work because the cells died. Then we tried an shRNA approach, which we had previous used in the lab for a number of glioma lines, and that didn’t work. The cells always started dying or they gained back STAT3. So like in the title, and as Debolina mentioned, it seems like the cancer stem cells are really addicted to the presence of STAT3.
I mean, I think that one of the future goals of our experiments, since we find out that STAT3 is so critical, is to work with some drug investigators to develop new STAT3 inhibitors that could be used to target glioblastoma. And one of the biggest problems in trying to develop a drug in glioblastoma is the fact that you have to get the drug to cross the blood brain barrier. And that was one of the issues of why, unfortunately, Debolina had to do all her animal experiments by subcutaneous injection, because it seemed like such a straightforward thing. But we could put the cells into the brain and we could establish the cell lines, but once we tried to knock down the expression of STAT3 by adding doxycycline, we couldn’t get enough doxycycline to cross the blood brain barrier. And it was a very frustrating time for poor Debolina.
We tried every possible way of delivering doxycycline to the mice. We tried it in the water, we tried it in the food, we tried it by oral gavage. We tried combinations of that and none of those ways worked. But we were able to do this when we did put the tumor into the flanks of the mice, we can knock it down. So, I mean, it doesn’t sound like a lot, but it was a major technical hurdle that she overcame with developing the dox inducible system, because the cells, since they require STAT3 so much, if you knock down STAT3, the cells just stop growing. So we had to come up with a way to study the mutants by putting them already into the cells and then knocking down the endogenous STAT3 so only the exogenous STAT3 was essentially being the one that was being expressed.
So it was technically a challenging project, but also, we did a lot of really sexy techniques. We did RNA-Seq in collaboration with Dr. Roussel over at St. Jude’s to analyze the genes that were regulated by STAT3. And some of the genes were kind of surprising. And I started out talking about how I started out working on interferon, and it was kind of interesting that STAT3, when you knock down STAT3, you turn on a number of interferon stimulated genes so it appears that STAT3 is playing a repressive role in the interferon response. And maybe that’s part of the importance of why STAT3 also regulates tumorigenesis. Interferon is an anti tumorigenic cytokine and STAT3 is turning off the interferon system because the interferon system is so anti tumorigenic and that’s a way that it promotes tumorigenesis. So there’s a lot of interesting data that this manuscript shows, and I think Debolina should be proud of herself during her time as a graduate student, that she was able to accomplish this.
Debolina Ganguly
Yeah. It took a long time, but I’m happy that it finally worked.Oncotarget | The critical role that STAT3 plays in glioma-initiating cells STAT3 addiction in glioma
—
Oncotarget is a unique platform designed to house scientific studies in a journal format that is available for anyone to read—without a paywall making access more difficult. This means information that has the potential to benefit our societies from the inside out can be shared with friends, neighbors, colleagues and other researchers, far and wide.
For media inquiries, please contact media@impactjournals.com.